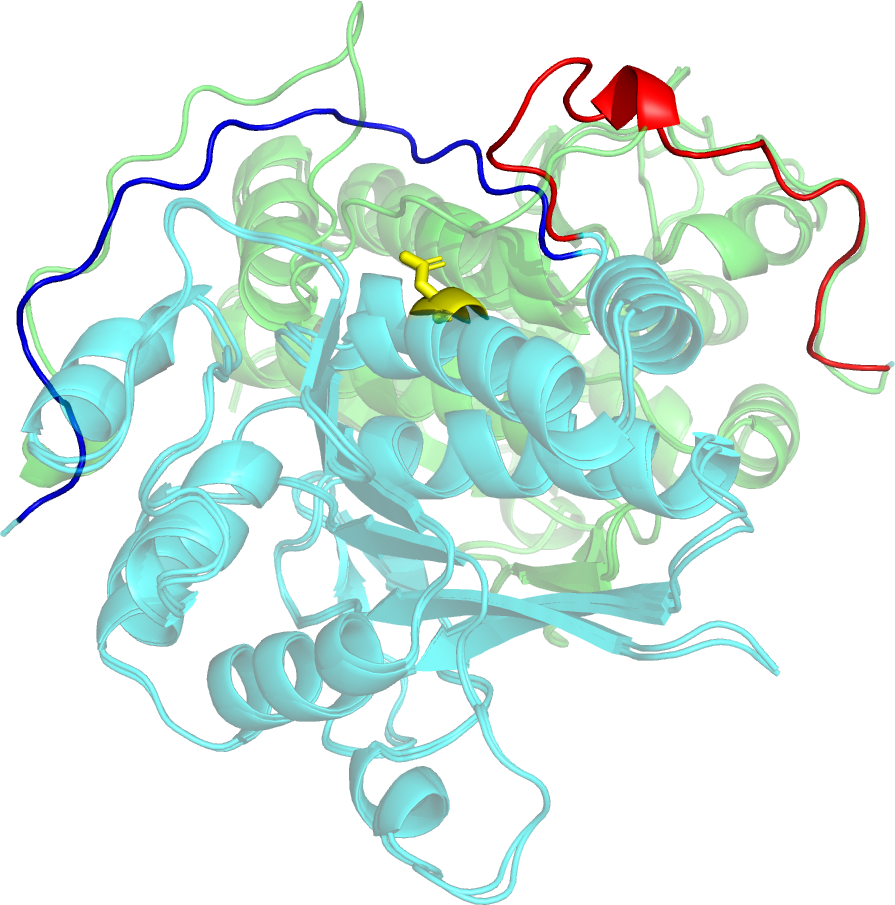
NMR studies can provide unique information about protein conformations in solution. In CASP14, three reference structures provided by solution NMR methods were available (T1027, T1029, and T1055), as well as a fourth data set of NMR-derived contacts for a integral membrane protein (T1088). For the three targets with NMR-based structures, the best prediction results ranged from very good (GDT_TS = 0.90, for T1055) to poor (GDT_TS = 0.47, for T1029). We explored the basis of these results by comparing all CASP14 prediction models against experimental NMR data. For T1027, the NMR data reveal extensive internal dynamics, presenting a unique challenge for protein structure prediction. The analysis of T1029 motivated exploration of a novel method of “inverse structure determination”, in which an AF2 model was used to guide NMR data analysis. NMR data provided to CASP predictor groups for target T1088, a 238-residue integral membrane porin, was also used to assess several NMR-assisted prediction methods. Most groups involved in this exercise generated similar beta-barrel models, with good agreement with the experimental data. However, as was also observed in CASP13, some pure prediction groups that did not use the NMR data generated structures for T1088 that better fit the NMR data than the models generated using these experimental data. These results demonstrate the remarkable power of modern methods to predict structures of proteins with accuracies rivaling solution NMR structures, and that it is now possible to reliably use prediction models to guide and complement experimental NMR data analysis.