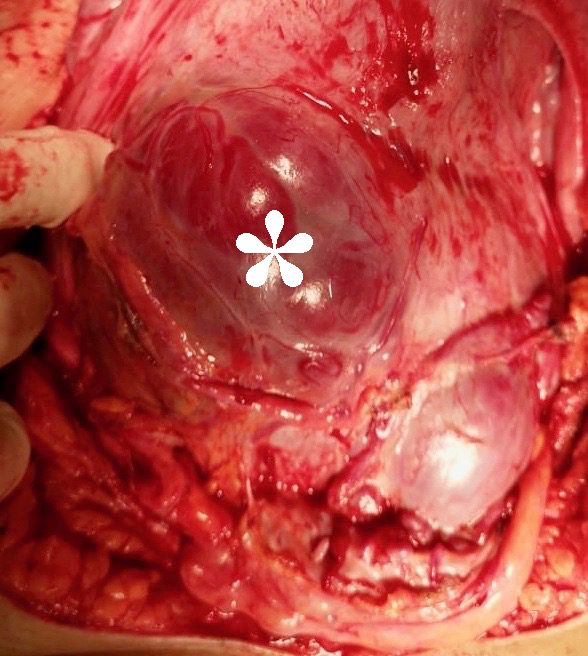
Placenta accreta spectrum: Welcome progress and a call for standardization.Mini-commentary for BJOG on Kayem et al 2021 BJOG-20-1462R3Placenta accreta spectrum (PAS) is among the most feared causes of maternal morbidity worldwide, and yet few prospective data are available to inform best practice. PAS is rare enough that rigorous study in a single center is difficult, but common enough that most obstetric hospitals now encounter PAS. Management and outcomes vary strikingly between hospitals and best practice, regrettably, is guided more by expert consensus than by level I evidence. In fact, most clinical questions regarding management of PAS are informed by essentially no prospective data (Collins et al. Am J Obstet Gynecol. 2019;220:511-526).Into this data void has come the PACCRETA cohort (Kayem et al. Act Obstet Gynecol Scand 2013;92:476-482) and some of its first results, published in this issue of BJOG (Kayem et al. BJOG 2021). PACCRETA is a prospective population-based study from 176 hospitals in France, capturing 30% of all French deliveries, from from 2013 to 2015. The study investigators identified 249, or 4.8 per 10,000, cases of PAS.Of all PAS patients, Kayem and colleagues found that a full half did not have the classic combination of risk factors for PAS (previa with history of cesarean). This group had lower morbidity and milder disease than those with the classic combination. Only 17% of those without classic risk factors were diagnosed antenatally. The message here is mixed: those without classic PAS risk factors are less likely to be diagnosed antenatally (bad) but appear to suffer less morbidity overall (good).But did these patients actually have PAS? Only 21% of those without prior cesarean and previa had a hysterectomy. Although this could be due to a regional preference for conservative treatments, the presence of false positives seems likely. Without a hysterectomy specimen, the diagnosis of PAS is difficult, controversial, and (in our opinion) highly susceptible to overdiagnosis. The authors define “strict” criteria for true cases of PAS, but several criteria depended entirely on the subjective assessment of a clinician faced with a difficult placental removal and the flawed principle of PAS as diagnosis of exclusion . Difficulty in manual placental removal or massive bleeding from an implantation site does not always indicate that microscopic PAS was present. Similarly, areas of prior cesarean section scar dehiscence (windows) where the placenta can be seen through the serosa are often diagnosed as percreta (Figure) without any histological evidence of the villous tissue having invaded through the serosa or beyond (Hecht et al. Modern Pathol 2020;33:2382-2396).We congratulate Kayem and colleagues for the current study and all of their important contributions to our understanding of PAS. However, these data illustrate the need for standardization of the definition of PAS, especially in conservatively managed cases with considerable potential for misdiagnosis. There is a desperate need for controlled studies of patients with antenatally suspected PAS with detailed and objective documentation of imaging, intra-operative findings, and when available, histopathological examination. In absence of such studies, the void of definitive data to guide treatment options will remain wide open.