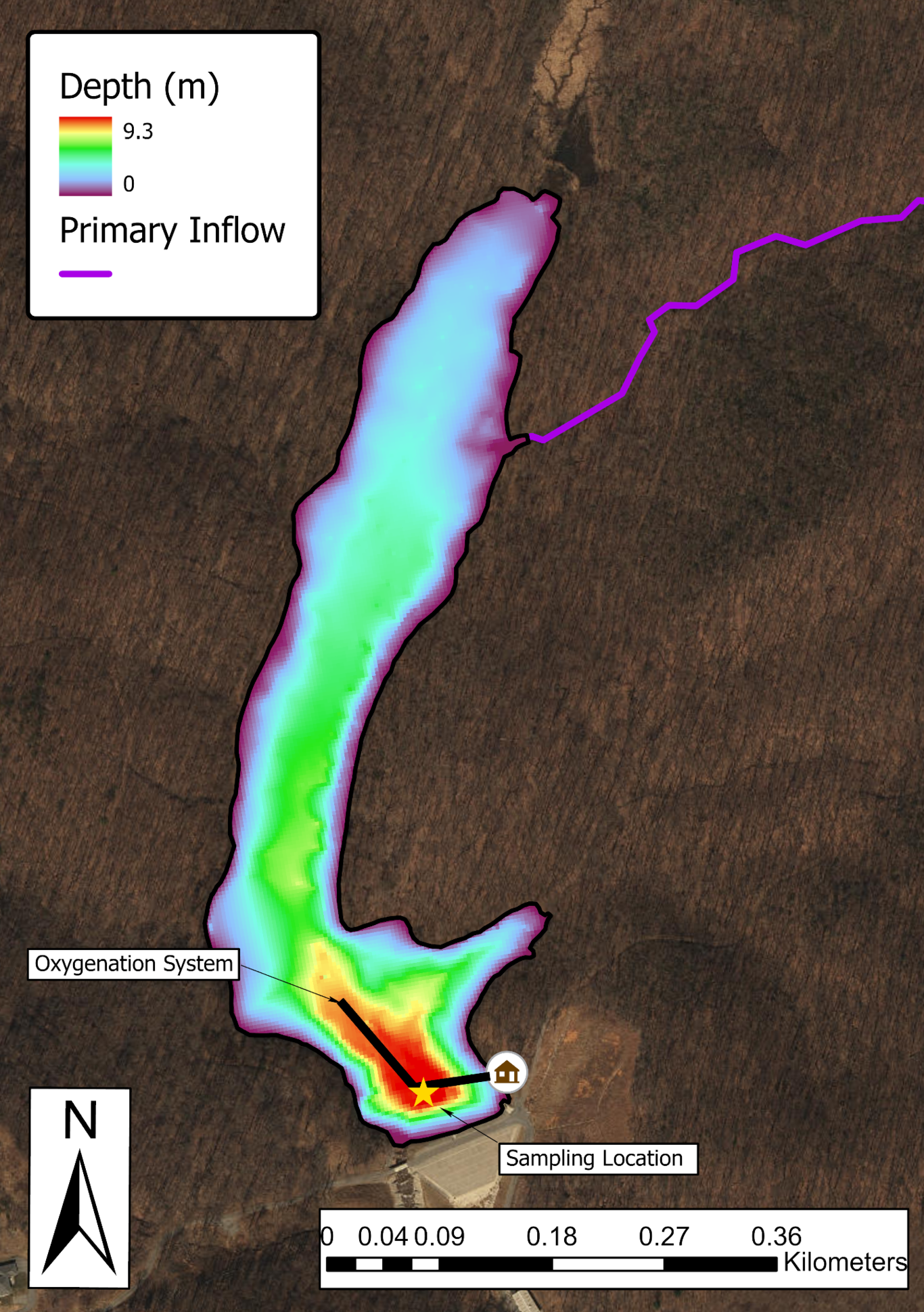
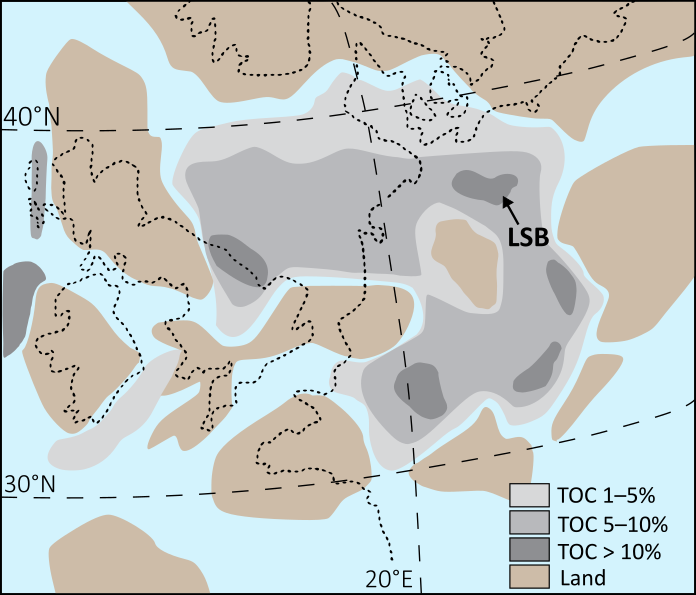
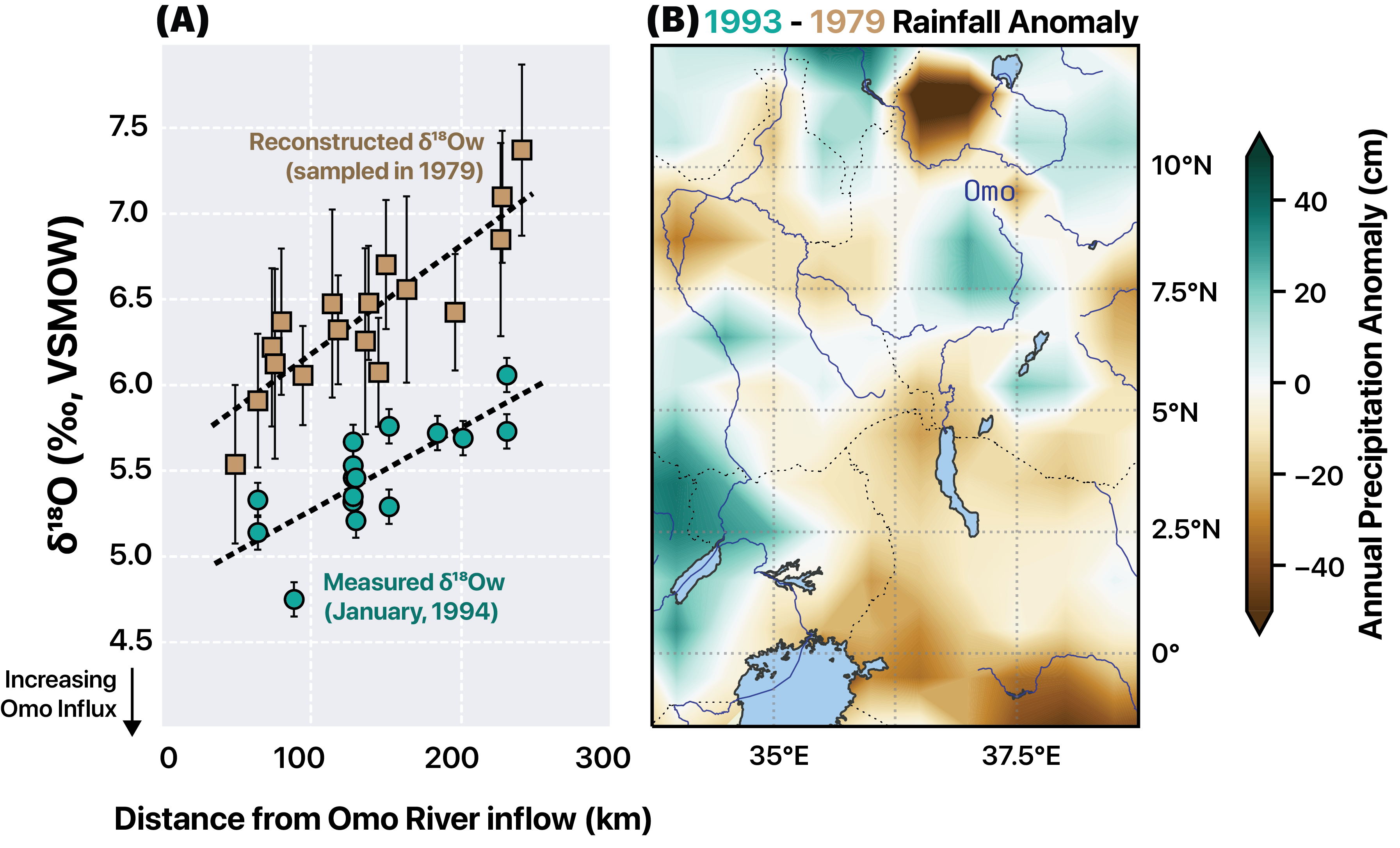
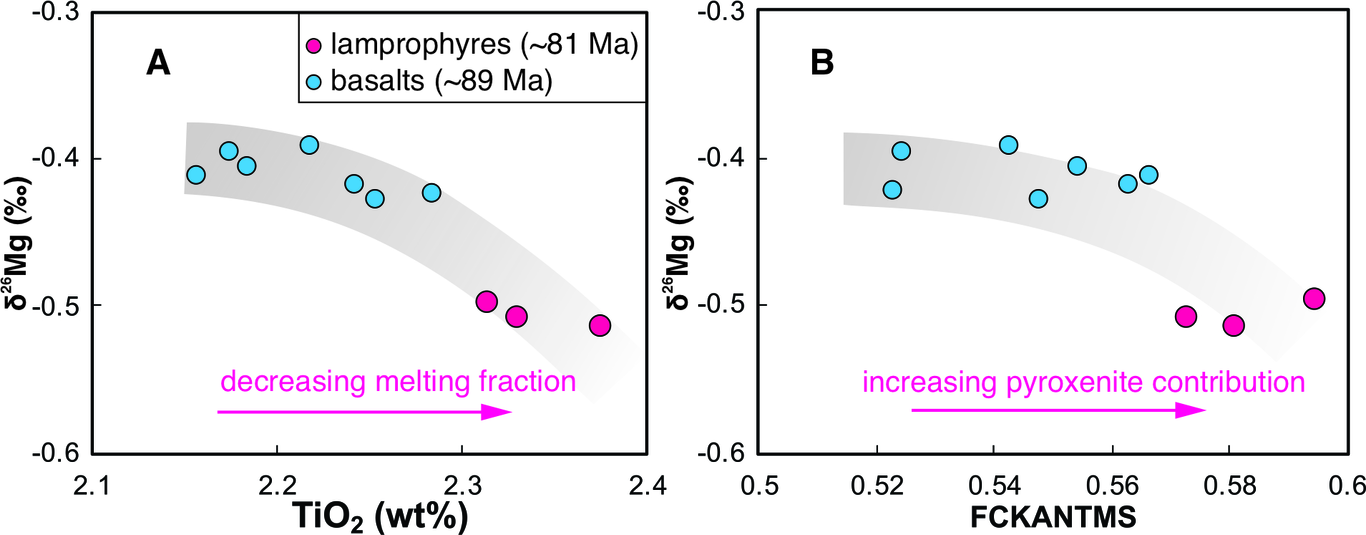
Moisture recycling via evapotranspiration (ET) is often invoked as a mechanism for the high deuterium excess signals observed in continental precipitation (dP). However, a global-scale analysis of precipitation monitoring station isotope data shows that metrics of ET contributions to precipitation (van der Ent et al., 2014) explain little dp variability on seasonal timescales. This occurs despite the fact that ET contributions increase by ~50% in continental locations such as the Eurasian interior from wet to dry seasons. To explain this apparent paradox, we hypothesize that the effects of ET on dP are dampened during dry seasons due to contributions from isotopically-evolved residual water storage that act to lower the d-excess of ET fluxes (dET), in combination with changes in transpiration fraction (T/ET). To test this hypothesis, we develop a parsimonious two-season (wet, dry) model for dET incorporating residual water storage and ET partitioning effects. We find that in environments with limited water storage, such as shallow-rooted grasslands, dry season dET is lower than wet season dET despite lower relative humidity. As global average ratios of annual water storage to precipitation are relatively low (Guntner et al., 2007), these dynamics may be widespread over continents. In environments where water storage is not limiting, such as groundwater-dependent ecosystems, dry season dET is still likely lower; however, this effect arises instead due to higher seasonal T/ET when energy-driven plant water use is enhanced and surface evaporation is relatively limited by water availability. Together, these analyses also indicate multiple mechanisms by which dET may be lower than dp during the same season, challenging the view that moisture recycling feedback increases the dp in continental interiors. This work demonstrates the potential complexity of seasonal dp dynamics and cautions against simple interpretations of dP as a process tracer for moisture recycling. References: Guntner et al., 2007. Water Resour. Res., 43, W05416. van der Ent et al., 2014. Earth Syst. Dynam., 5, 471–489.