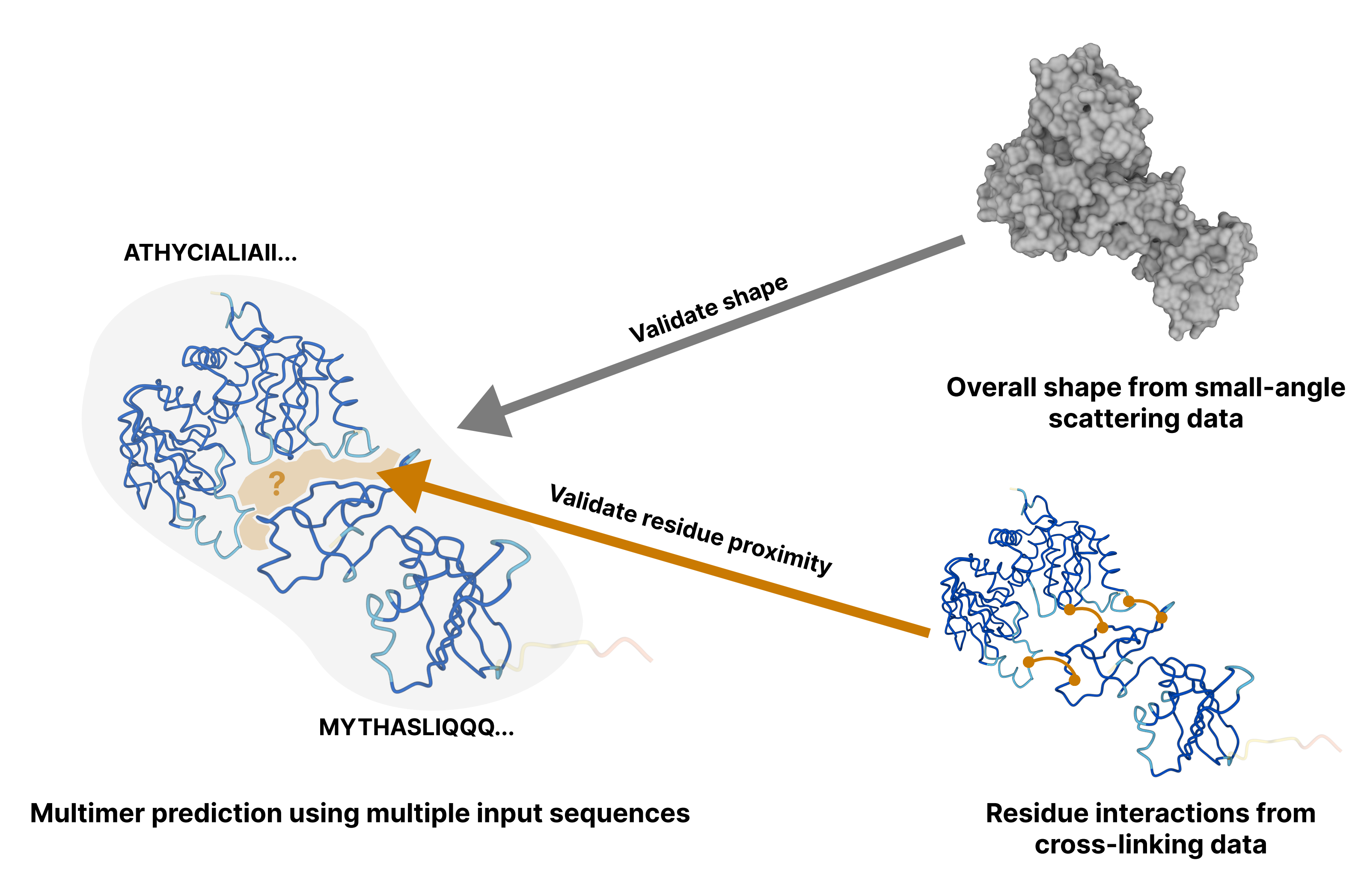
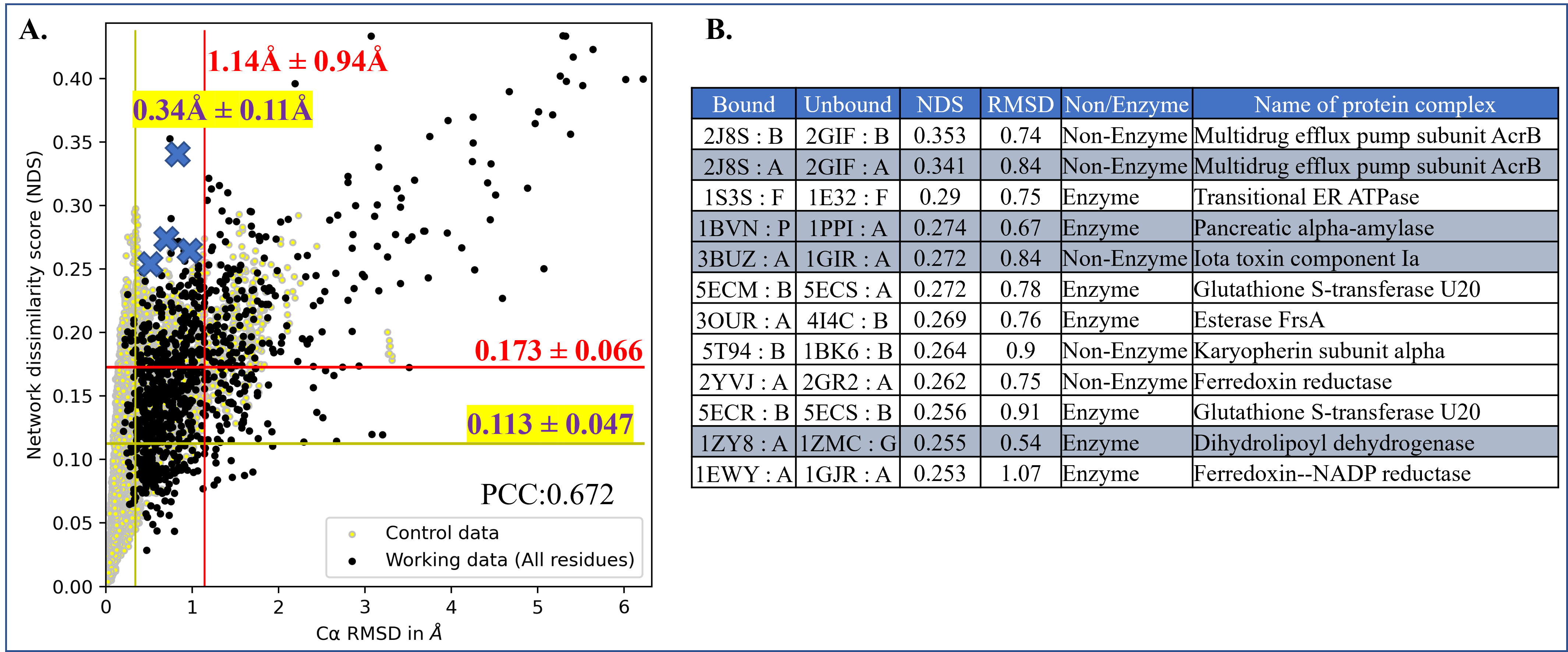
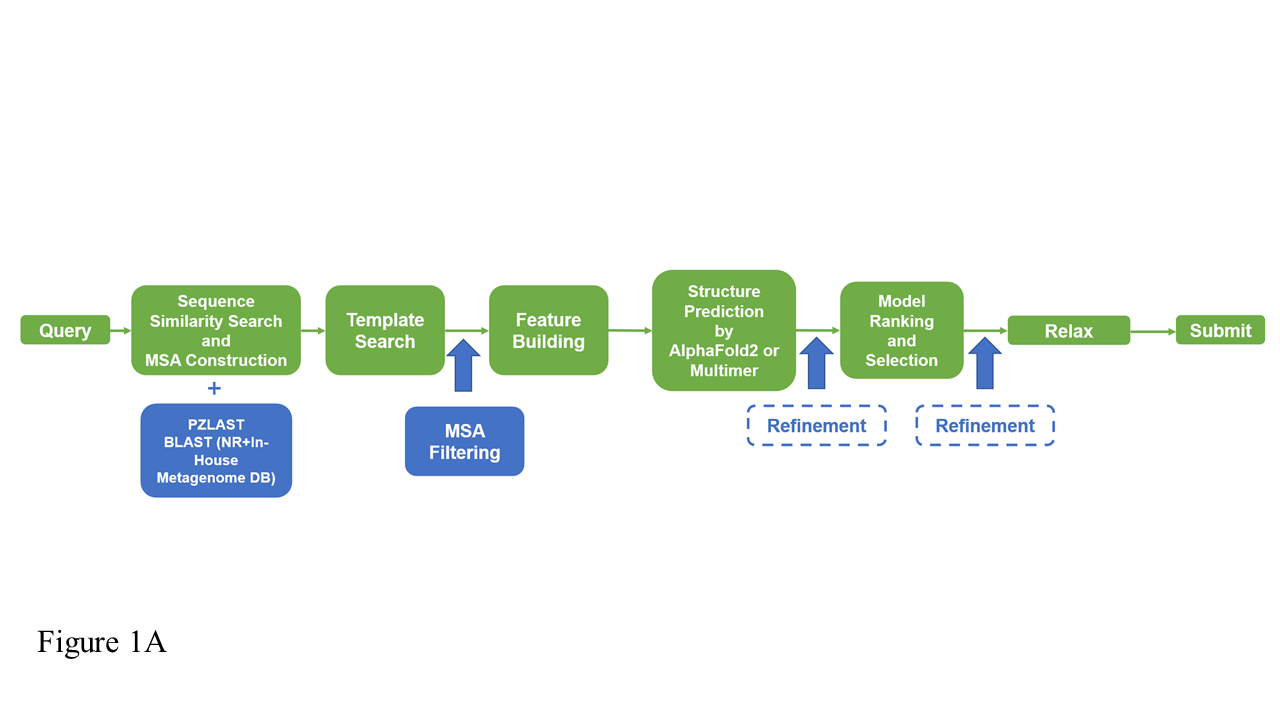
Computing protein structure from amino acid sequence information has been a long-standing grand challenge. CASP (Critical Assessment of Structure Prediction) conducts community experiments aimed at advancing solutions to this and related problems. Experiments are conducted every two years. The 2020 experiment (CASP14) saw major progress, with the second generation of deep learning methods delivering accuracy comparable with experiment for many single proteins. There is an expectation that these methods will have much wider application in computational structural biology. Here we summarize results from the most recent experiment, CASP15, in 2022, with an emphasis on new deep learning-driven progress. Other papers in this special issue of Proteins provide more detailed analysis. For single protein structures, the AlphaFold2 deep learning method is still superior to other approaches, but there are two points of note. First, although AlphaFold2 was the core of all the most successful methods, there was a wide variety of implementation and combination with other methods. Second, using the standard AlphaFold2 protocol and default parameters only produces the highest quality result for about two thirds of the targets, and more extensive sampling is required for the others. The major advance in this CASP is the enormous increase in the accuracy of computed protein complexes, achieved by the use of deep learning methods, although overall these do not fully match the performance for single proteins. Here too, AlphaFold2 based method perform best, and again more extensive sampling than the defaults is often required. Also of note are the encouraging early results on the use of deep learning to compute ensembles of macromolecular structures. Critically for the usability of computed structures, for both single proteins and protein complexes, deep learning derived estimates of both local and global accuracy are of high quality, however the estimates in interface regions are slightly less reliable. CASP15 also included computation of RNA structures for the first time. Here, the classical approaches produced better agreement with experiment than the new deep learning ones, and accuracy is limited. Also, for the first time, CASP included the computation of protein-ligand complexes, an area of special interest for drug design. Here too, classical methods were still superior to deep learning ones. Many new approaches were discussed at the CASP conference, and it is clear methods will continue to advance.